


Approximately 85%–90% of individuals develop acne during adolescence, a time period that coincides with the onset and transition through puberty.
It is well established that the physical and biochemical properties of the skin are influenced by puberty and skin colonization by microbes. Functional genes encoded in the microbes may alter skin ecology as well.
In order to gain a better understanding of the microbiome-host interactions in acne, a multi-omics comparison of facial skin microbiome data was conducted between individuals with and without active acne during puberty.
Read on to learn more about this study and its findings – and how the CosmosID-Hub played a key role in this research.
Background
Previous studies have indicated that the skin microbiome plays an important role in the development of acne. This suggests that the skin microbiota composition and metabolic activity may be different between individuals with and without active acne during puberty.
To investigate this further, a multi-omics comparison was conducted to determine if there are any meaningful differences between facial skin microbiome data from individuals with and without active acne during puberty.
Our analysis
To compare and contrast skin microbiome compositions of acne patients and healthy controls, we analyzed 48 skin microbiome samples (27 acne skin and 21 healthy skin samples), from the study of Schneider et al. (2022) with the NCBI Sequence Read Archive accession ID PRJNA856701.
We used the CosmosID-HUB Microbiome for accurate profiling of the bacterial and fungal species relative abundances among healthy and acne skin samples along with prediction of functional pathways.
To understand if a distinct skin microbiome and associated functional metagenome emerges in paediatric acne subjects, we comparatively analyzed the facial skin microbiome of children from 7 to 17 years old.
We identified skin microbiome alterations in bacteria and fungi associated with acne skin, as well as functional metagenomic differences. Altogether, functional metagenomic variations mirror the microbiome variation in acne skin.
Findings
Species richness and evenness
To illustrate the variation in bacterial species richness and evenness, we calculated the Shannon’s index of diversity between the two cohorts using a Wilcoxon rank sum test.
The findings of the Wilcoxon test illustrated a significant difference in alpha diversity when species richness and evenness are considered. (Wilcoxon Test p-value < 0.05, Fig. 1).
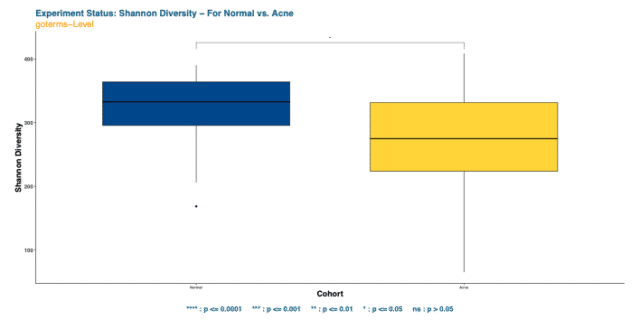
The finding of the Shannon’s index diversity comparison suggests that the skin microbiome of children with acne has significantly lower diversity than children with normal skin.
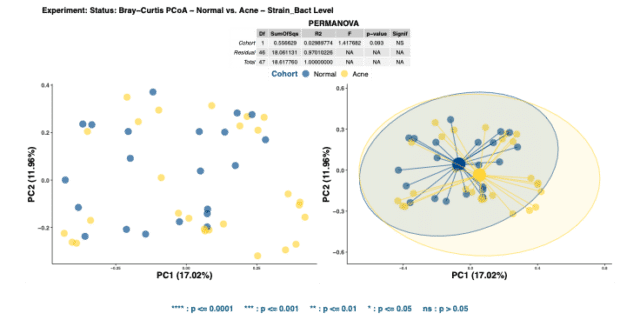
The significance in alpha diversity between the two cohorts suggests that species richness and evenness is limited in the skin microbiome of acne patients. However, comparing the beta diversities of acne and healthy skin via Bray-Curtis dissimilarity matrices illustrates no significant dissimilarity in bacterial compositions (Figure 2).
No significant beta diversity findings implies that the bacterial community compositions between the groups do not differ from each other. Repeating the same analysis to detect dissimilarity in mycobiome compositions also yields insignificant results.
Functional pathways of microbial communities
After observing significant differences in bacterial sp
ecies richness and evenness, but no significant dissimilarity in microbiome or mycobiome community composition, we questioned whether the microbial communities from the two skin cohorts were encoding different functional pathways.
To dissect that, we first compared the Shannon’s Diversity index of Gene Ontology (GO) terms present in healthy and acne skin samples. The findings illustra
ted that the functional pathways in GO terms are significantly more limited regarding pathway term richness and evenness in acne skin when compared to the healthy (see Figure 3).
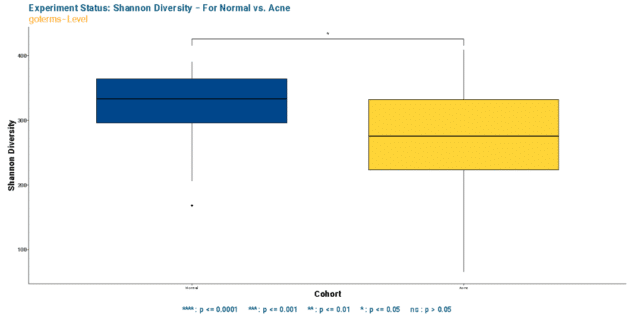
Observing a significant difference in alpha diversity raised the question of whether the dissimilarity persisted in fuctional GO term relative abundance composition.
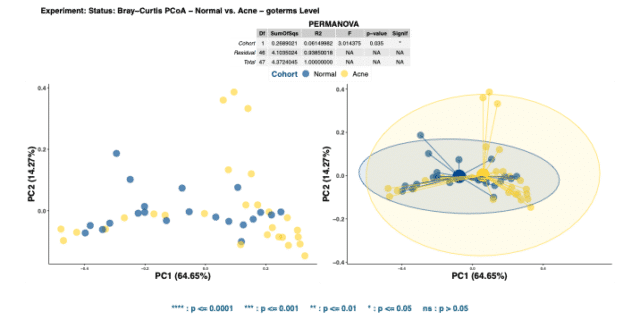
Comparing the Bray-Curtis dissimilarity matrices of GO terms confirmed the functional pathways encoded in the two cohorts were indeed significantly dissimilar (Figure 4). Differences in functions present in different skin types may drive their bio-ecological differences.
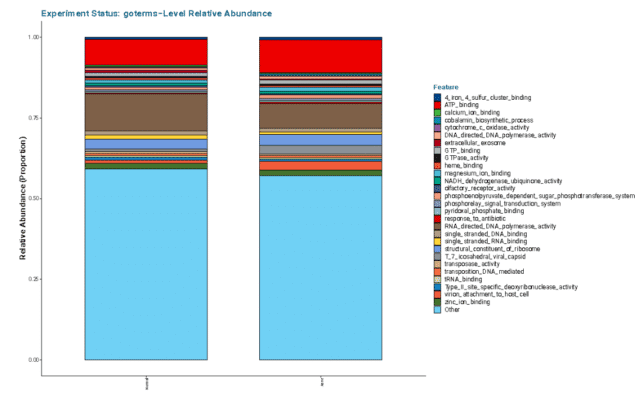
Altogether, the relative abundance of GO terms illustrated that acne facial skin has lower mean relative abundance of RNA-Directed DNA Polymerase (see Figure 5) highlighting that there may be less DNA production and replication in acne skin.
Genetic correlation
Examining relative abundances of bacterial, fungal and functional GO terms in healthy and acne facial skin led to the question of whether these are correlated. Answering this question could illustrate if certain genetic functions are consistently associated with the presence and relative abundance of specific subsets of species or if species interact with each other in different skin ecologies, such as acne status.
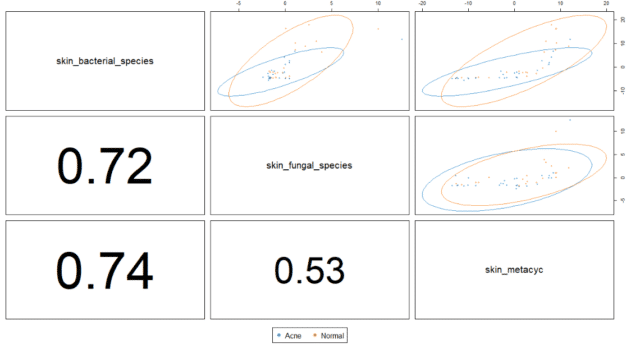
To investigate bacterial, fungal and functional GO term correlations, we predicted the extent of discriminatory potential of features between the cohorts using a sparse Partial Least Squares – Discriminant Analysis (sPLS-DA). Next, we examined the relationships between them using Data Integration Analysis for Biomarker discovery using Latent variable approaches for Omics studies (DIABLO).
Analyzing the correlations between the different types of data illustrated that bacterial species strongly correlated with fungal species and functional GO terms. However, the correlation between fungal species and functional GO terms remained limited when compared to that of bacterial species (see Figure 6).
After having observed strong correlations of bacterial species with fungal species and GO terms, the next step was investigating the direction of the relationship for different taxonomic and functional terms.
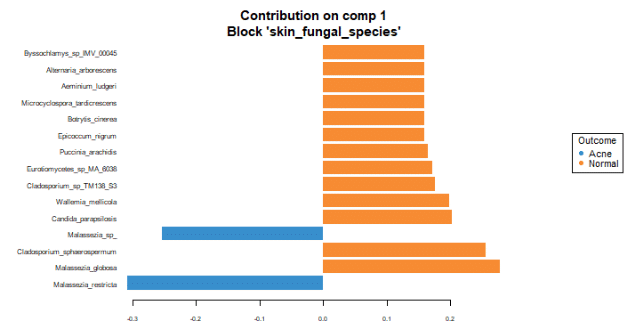
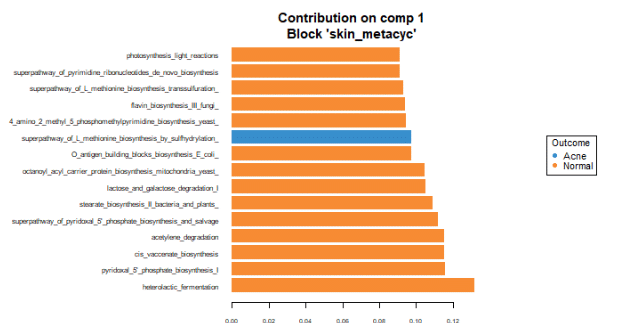
To identify the intensity and direction of the relationship for the terms that explained the most variance, we illustrated the loadings of the top terms in the top variate for bacterial (Figure 7), fungal species (Figure 8) and GO terms (Figure 9).
Bacterial variance
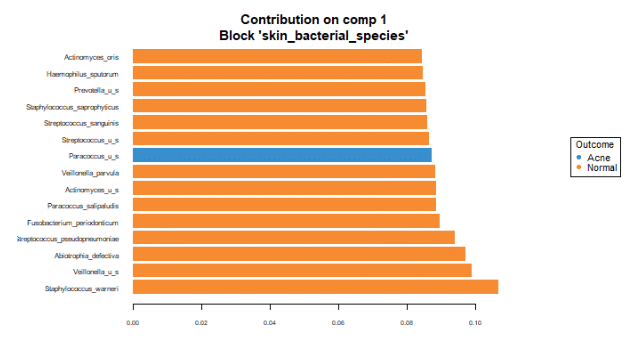
The loading graphs suggest that Staphylococcus warneri is most important in explaining the bacterial variance in component 1, being more prevalent in normal skin.
Malassezia restricta is the most important fungal species for component 1. It is most prevalent in skin with acne. It inversely correlates with other top loadings in the first variate, with the exception of another Malassezia sp. which inversely correlates with other loadings in variate 1. Regarding GO terms, heterolactic fermentation is the most important functional pathway in component 1 and is prevalent in normal skin.
Overall, there are more taxonomic and GO terms whose loadings explain more variation and are associated with normal skin when compared to the loadings that are prevalent in acne skin.
These findings suggest that with acne, the skin may lose species diversity and functional pathways. Among detected fungi, specific Malassezia species are prevalent in healthy skin (Malassezia globosa), and in skin with acne (Malassezia restricta and Malassezia sp). These associations suggest differing roles of these species in the presence of acne.
After observing differences in correlations of taxonomic and functional terms in acne and healthy skin, we examined how individual terms correlated with each other.
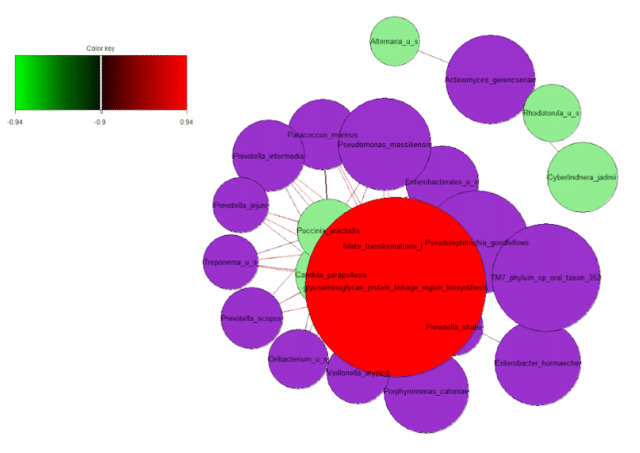
To illustrate this, we plotted a network graph using the correlations of bacterial, fungal and GO terms (Figure 10). To illustrate the top correlations in a concise fashion, we selected an arbitrary threshold of r > 0.94 to visualize the relationships. The network graph illustrated two distinct groups of correlations.
The first cluster consisted of strong correlations of the bacterium Actinomyces gerencseriae, a normal skin commensal that may opportunistically cause hip prosthesis infection, with fungi Alternaria which may cause skin infections, Rhodotorula, which can disrupt the skin barrier and Cyberlindnera jadinii, another commensal skin fungi that could cause opportunistic infections. In the second group, we observed correlations of taxa and functions involving folate and glycosaminoglycan production and processes.
Both folates and glycosaminoglycans hydrate and rejuvenate the skin, and glycosaminoglycans also help maintain its elasticity and volume. This suggests that acne skin may lack of hydration and anti-aging functions, which could be associated with the increased prevalence of fungal taxa.
Summary: what does this mean?
Our comparison of taxonomic and functional metagenomics of acne and healthy facial skin illustrated that bacterial and functional alpha diversity is significantly reduced in skin with acne.
Moreover, there are differences between acne and healthy skin in their functional potential. Altogether, these findings may serve as biomarkers to treat acne during puberty or predict acne development through examining functional and taxonomic variations in facial skin.
These results suggest that restoring balance to facial skin microbiota through targeting specific species and functions may aid in the treatment of acne. Specific targeting of Malassezia restricta and supplementation of acne-prone skin with folate and glycosaminoglycan may be beneficial. These treatments may be especially important for teenagers during puberty, when hormonal changes can trigger an increase in sebum production and lead to new outbreaks of acne.
How did the CosmosID-HUB support this research?
The CosmosID-HUB supported this research by rapidly identifying the taxonomic and functional differences between acne and healthy facial skin. By using metagenomics data from samples taken from different individuals, CosmosID’s innovative technology allowed us to accurately compare them in order to identify the key correlative taxa that distinguished these two groups.
In addition, CosmosID’s fast, cost-effective and reliable microbial identification system enabled us to identify the key genera and species associated with acne. This data provided evidence of a relationship between Malassezia interplay and acne status, giving us insight into possible treatments and clinical implications for managing skin health.
By providing an accurate analysis of the microbiome data, CosmosID has supported the development of novel insights into the microbiome associated with acne and provided evidence to guide efforts for managing skin health.
Looking for support with your next research project?
If you’re looking for support with your next research project, CosmosID offers a wide range of microbial sequencing, identification and analysis services. Our comprehensive platform can provide you with results in minutes, enabling more efficient and cost-effective data analysis.
With advanced taxonomic and functional insights into the microbiome, we are committed to helping scientists make discoveries that will advance human health and well-being.
