

Even after the height of the pandemic, there is still an ongoing battle against the global coronavirus disease, meaning the realm of metagenomics and metatranscriptomics continues to play a pivotal role in unraveling the complexities of the SARS-CoV-2 virus.
As of 2023, scientists persistently engage in collaborative efforts to harness metagenomic and metatranscriptomic data for comprehensive insights into the viral genome, transmission dynamics, and the intricate interplay between the virus and its host.
At the forefront of these endeavors is CosmosID, diligently monitoring the outbreak and integrating cutting-edge RNA-sequencing-based data into their analysis platform.
The urgency stems from the gravity of the public health threat posed by SARS-CoV-2, a highly transmissible coronavirus responsible for the ongoing global health crisis.
In this expert guide, we’ll share our metagenomic and metatranscriptomic insights into SARS-CoV-2, COVID-19, and how metagenomic and metatranscriptomic analysis can aid in the detection, surveillance, and management of this novel coronavirus.
Highlights
- Emergence of SARS-CoV-2 caused a pandemic and global health biothreat in 2020.
- SARS-CoV-2 is spread by human-to-human transmission via respiratory droplets or direct contact.
- Monitoring and controlling infection to prevent spread of SARS-CoV-2 constituted primary intervention in 2020.
- In 2023, metagenomic and metatranscriptomic analysis remains critical to understanding the evolutionary dynamics and transmission patterns of SARS-CoV-2.
- Moreover, it can provide crucial insights into host-pathogen interactions, drug resistance, and potential diagnostic markers for COVID-19.
- CosmosID accurately detects SARS-CoV-2 in samples through metatranscriptomics.
- Researchers can upload metagenomic and metatranscriptomic sequence files to CosmosID for SARS-CoV-2 identification and characterization.
What is SARS-CoV-2?
What is SARS-CoV-2?
SARS-CoV-2 is the terminology for “Severe Acute Respiratory Syndrome Coronavirus 2”, commonly referred to as “Coronavirus”. This pathogenic virus causes coronavirus disease (COVID-19) and belongs to a family of single-stranded RNA viruses. The virus genome spans 29,891 nucleotides. This type of virus can be found in many animal species and and has the ability to cross the animal species barrier and infect humans. When its genomic sequence became available, scientists compared this virus genome with other available coronavirus genomes. It was concluded that a novel virus with the closest known relatives being SARS-CoV, the virus causing the SARS outbreak in 2003, and coronavirus carried by bats.
How Has Our Understanding of COVID-19 Changed From 2020-2024?
Since the beginning of the pandemic, our understanding of COVID-19 has evolved significantly. In 2020, there was limited knowledge about the virus and its transmission patterns. However, as more data became available through metagenomic and metatranscriptomic analysis, scientists were able to gain a deeper understanding of the virus and its impact on human health.
One major change in our understanding is the recognition that SARS-CoV-2 is primarily spread through respiratory droplets and direct contact with infected individuals. This understanding led to the implementation of measures such as wearing masks and social distancing to prevent transmission.
Additionally, research has revealed that COVID-19 can cause a wide range of symptoms, from mild to severe, and can affect multiple organ systems in the body. This has highlighted the need for comprehensive metagenomic and metatranscriptomic analysis to better understand how the virus interacts with the human body.
Furthermore, as more people have been vaccinated and recovered from COVID-19, researchers have been able to gather valuable data on the immune response to the severe disease. This has led to a greater understanding of potential treatment options and the development of effective vaccines.
Overall, our understanding of COVID-19 has greatly improved since 2020, but ongoing metagenomic and metatranscriptomic analysis remains crucial in furthering our knowledge and aiding in the fight against this virus.
COVID-19, Metagenomics + Metatranscriptomics, and the Microbiome: The Basics
In relation to metagenomics and metatranscriptomics, links have been demonstrated between COVID-19 and certain gut microbes. Below, we’ll examine the relationship between COVID-19 and the microbiome, including how metagenomics and metatranscriptomics are used to study these connections.
Gut Microbiota and SARS-CoV-2
In recent research, significant alterations in the gut microbiota composition have been associated with SARS-CoV-2 infection, revealing a complex interplay between the virus and the host’s intestinal microbiota.
Gut microbial taxa, particularly beneficial species, appear to decline in the presence of the SARS-CoV-2 virus, indicating a dysbiotic state.
In critically ill patients with COVID-19, this dysbiosis is often exacerbated, resulting in a gut microbiota composition that could potentially reinforce the severity of the infection. Specifically, studies have shown significant correlations between the depletion of certain microbial species and the inflammatory responses observed in these patients.
Moreover, the altered gut microbiota composition in COVID-19 patients is associated with distinct clinical characteristics, including disease severity and immune response. As such, modulating gut microbial communities could be a promising line of research for COVID-19, as well as other inflammatory diseases, such as IBS and Celiac Disease.
It is important to note that these findings underscore the need for further investigations into the complex interactions between SARS-CoV-2 and the gut microbiota. Understanding these associations could provide us with novel therapeutic strategies for managing COVID-19, such as microbiota-based interventions.
Metagenomic & Metatranscriptomic Analysis and SARS-CoV-2
Metagenomic & Metatranscriptomic analysis has proven instrumental in understanding the SARS-CoV-2 virus, particularly in discerning its interaction with the host’s gut microbiota. The gut microbial ecology influences the host’s immune response to the virus, potentially exacerbating the severity of COVID-19 symptoms.
An imbalanced gut microbiota, or dysbiosis, can lead to an aberrant immune response, resulting in uncontrolled inflammation and subsequent development of acute respiratory failure, a lethal manifestation of severe COVID-19.
Moreover, metagenomics and metatranscriptomics also facilitates the detection and surveillance of SARS-CoV-2 variants by enabling genomic characterization. As a result, tailored treatments can be developed to combat specific virus strains.
However, it’s crucial that we continue to explore the gut-SARS-CoV-2 axis, as enhancing our knowledge on how gut microbial ecology influences disease progression could potentially transform our approach to managing and treating COVID-19.
In the future, modulating gut microbiota might serve as a viable strategy to both reduce the severity of COVID-19 and prevent acute respiratory failure.
Unlock the power of the microbiome with CosmosID. Get in touch today.
Automated placement of SARS-CoV-2 to closest known viruses
On January 22, 2020 we downloaded the novel coronavirus genome (now called SARS-CoV-2) and analyzed it using the CosmosID metagenomics analysis platform. We had not yet added this new genome to our database and wanted to see which genomes, if any, our algorithms would find as closest matches. The results we obtained within minutes correspond with the phylogenetic research being done by the genomics community.
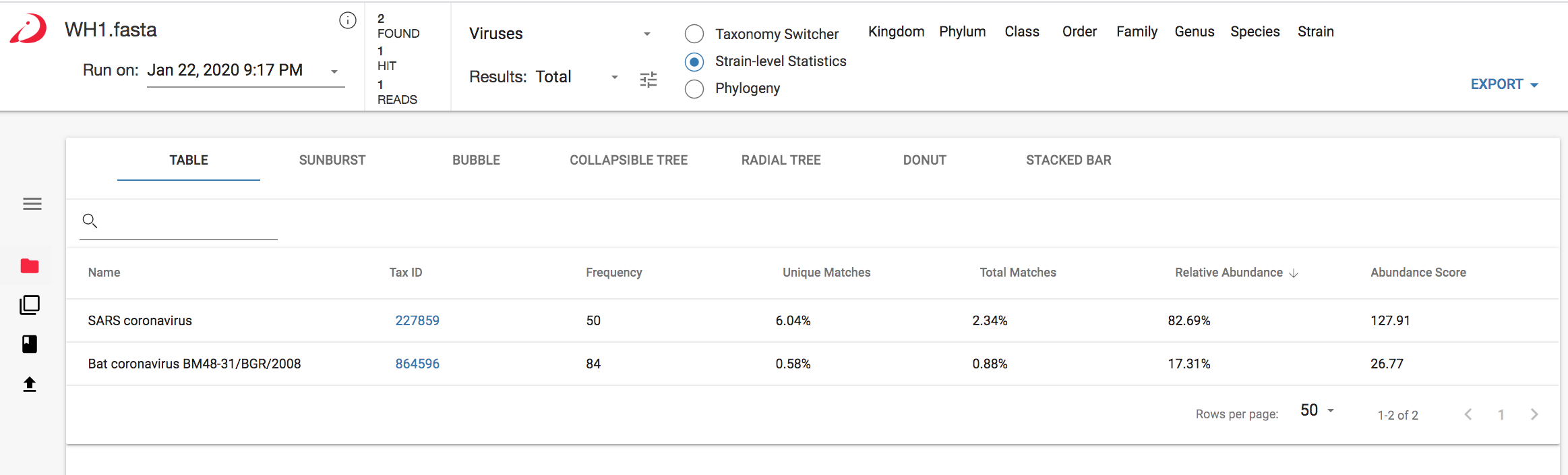
Figure 1 CosmosID results for closest matches to the SARS-CoV-2 genome before adding it to the CosmosID database (analysis on app.cosmosid.com)
As you can see in Figure 1 above, the CosmosID platform detected SARS coronavirus, at the species level, and Bat coronavirus BM48-31/BGR/2008 to strain-level identification. The platform compares NGS reads to sequence signatures (i.e., kmers) in a database arranged as a phylogenetic tree, and contains unique and shared kmers that map to each level in the tree. Within minutes the fully automated analysis identified kmers that pointed to the same bat coronavirus (highlighted in Figure 2 below) that Zhou et al. (2020) had identified as phylogenetically similar to SARS-CoV-2.

Figure 2 Phylogenetic tree from Zhou et al. (2020) showing the placement of five strains of 2019-nCov (original nomenclature for SARS-CoV-2). The arrow and highlighted name highlight the virus that was chosen as the closest strain by CosmosID automated analysis in the cloud.
This example demonstrates that the unique phylogenetic structure of the CosmosID database even allows a meaningful classification of pathogens that at the point of analysis were still unknown to the world (and therefore the database). Nature published the findings by Zhou et al. (2020) the day after CosmosID concluded our analysis.
Detecting SARS-CoV-2 in Metagenomic and Metatranscriptomic Samples
The SARS-CoV-2 genome is now in the CosmosID database. As the virus continues to spread, it is becoming critical to detect and classify the virus in patient samples so that individuals suspected of carrying the disease can be identified. While many labs around the world are using RT-PCR to detect SARS-CoV-2, a more precise method of detection is sequencing RNA in patient samples. A potential limitation of this method is that only a small percentage of the reads may include the virus of interest, making it potentially more difficult to identify. The upside of metagenomic sequencing on the other hand is the method’s ability to readily detect secondary pathogens that patients infected with SARS-CoV-2 may have acquired.
To assess the performance of CosmosID using metagenomic samples from patients diagnosed with COVID-19, our team analyzed nine bronchoalveolar lavage (BAL) metagenomic samples (deposited in the NCBI Sequence Read Archive under https://www.ncbi.nlm.nih.gov/biosample/SAMN14082199) through the CosmosID cloud application after we had included the SARS-CoV-2 genome to the CosmosID viral database.

Figure 3 Number of reads per sample from BAL metagenomic samples (analysis on app.cosmosid.com)
Despite the fact that we challenged our metagenomic analysis platform with samples that contained in addition to the coronavirus also the microbial background associated with BAL samples, and despite several cases of shallow sequencing depth (of 5M reads or less), we were able to identify SARS-CoV-2 in all of the samples. In addition, as you’d expect when using metagenomic sequencing, the platform reported other bacteria and viruses found in the respiratory samples as shown in the heat map in Figure 4.

Figure 4 Heat map of viruses and phages detected in COVID-19 patient BAL samples, including SARS-CoV-2, with a box around the name (analysis on app.cosmosid.com)
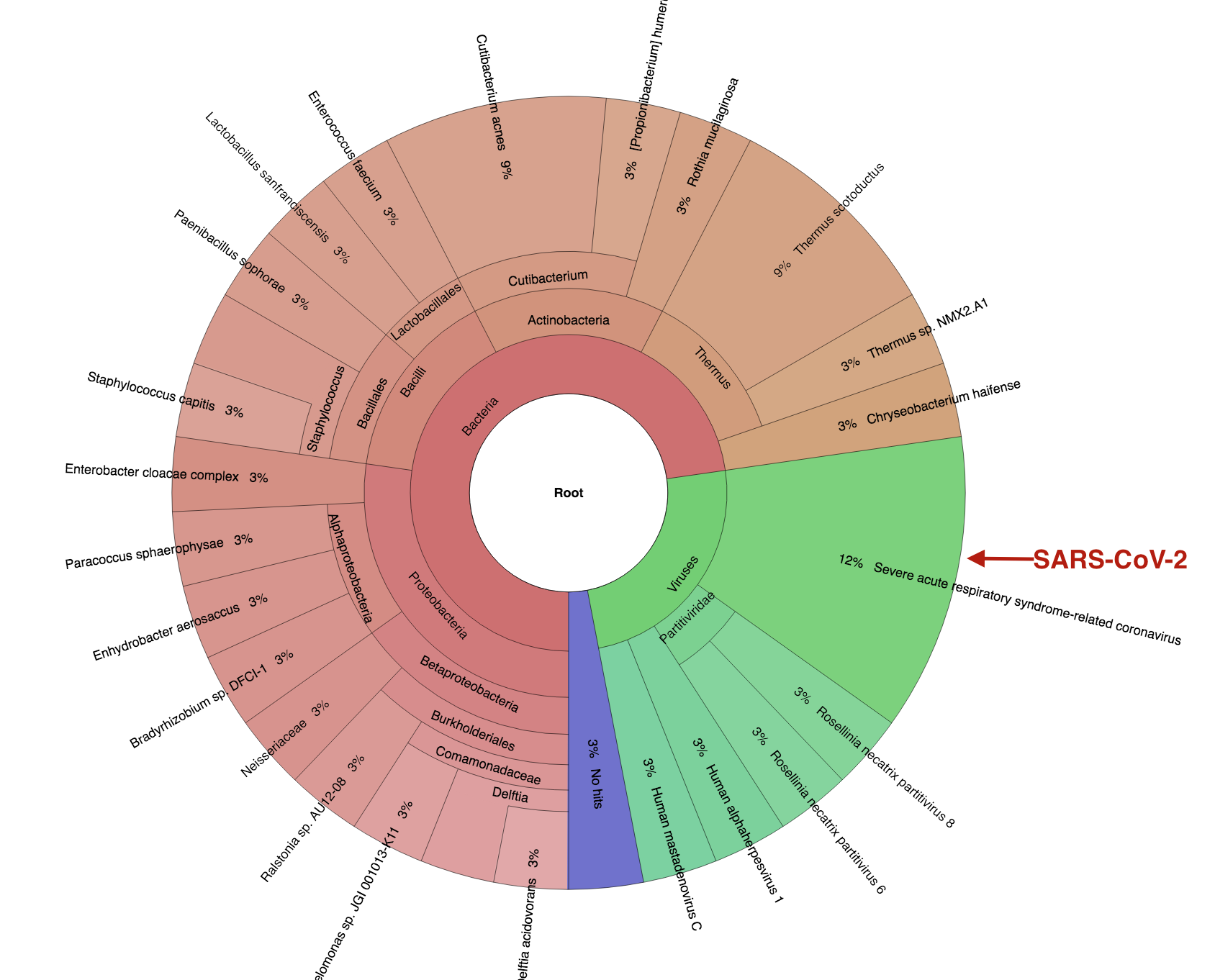
Figure 5 Krona plot showing bacteria and viruses in a single COVID-19 patient BAL metagenomic sample (analysis from app.cosmosid.com)
How To Run Your Metagenomic and Metatranscriptomic Samples
CosmosID can detect and identify SARS-CoV-2 in samples using metagenomic analysis. Researchers may use the CosmosID platform for this analysis. CosmosID does not provide diagnostic tests, but the CosmosID application is highly suitable for research purposes. Please reach out to us at info@cosmosid.com to learn more. Our team is eager to work with the community to achieve better understanding and to help contain the virus. We need to work together so that our lives can return back to normal.
Recommended End to End Workflow
Collect Sample → RNA Extraction ((QIAGEN QIAmp Viral Mini Kit, PN 52904) → RiboZero Gold rRNA depletion protocol to remove human cytoplasmic and mitochondrial rRNA (Illumina, 48 samples, Cat no. 20020598, 96 samples, 20020599) → Sequencing-ready library preparation using the TruSeq Stranded Total RNA Library Prep Gold kit (Illumina, Cat no. 20020599) along with the IDT for Illumina TruSeq RNA UD Indexes (96 indexes, 96 samples) (Illumina, Cat no. 20022371). RNA fragmentation, first- and second-strand cDNA synthesis, adenylation, adapter ligation, and amplification, according to the TruSeq Stranded Total RNA protocol. After amplification, the prepared libraries should be quantified, pooled, and loaded onto your preferred DNA Sequencer. We recommend sequencing reads at least 75bp in length. → Analyze sequence data on app.cosmosid.com
Unlock the power of the microbiome with CosmosID. Get in touch today.
CosmosID Recommended End-to-End Workflow For COVID-19 Metagenomics
The recommended end-to-end workflow involves several steps and actions to be performed. The workflow can be summarized as follows:
- Collect Sample: This step involves the collection of samples for analysis. It could be biological samples, environmental samples, or any other relevant samples required for the analysis.
- RNA Extraction: After sample collection, RNA extraction is performed. This process isolates and purifies the RNA molecules from the collected samples. The QIAGEN QIAmp Viral RNA Mini Kit is used for this purpose.
- Transcription: Once RNA extraction is complete, the next step is transcription. This process involves converting the extracted RNA into complementary DNA (cDNA) using reverse transcription methods.
- Library Preparation: After transcription, library preparation is carried out. This process involves preparing the DNA library for sequencing or other downstream analyses. Library preparation includes steps such as DNA fragmenting, end repair, adapter ligation, and PCR amplification.
- Sequencing: Once the library is prepared, sequencing is performed. This step involves determining the order of nucleotides in the DNA fragments. Various sequencing methods, such as Next-Generation Sequencing (NGS), can be employed.
- Data Analysis: Once the sequencing is completed, data analysis is conducted. This involves analyzing the sequence data and deriving meaningful insights from it using bioinformatics tools. These insights can help researchers understand biological processes and gain new knowledge about living organisms.
- Visualization: To gain insights from the analyzed data, visualization is often employed. This can include generating charts, graphs, or plots that represent the data in a visually understandable manner. Visualization can help in discovering patterns, trends, and relationships within the data.
The recommended end-to-end workflow involves these steps to successfully analyze and interpret data from sample collection to visualization. Please note that the workflow may vary depending on the specific context and requirements of the analysis.
COVID-19 Metagenomics & Metatranscriptomics: Closing Thoughts
The recommended end-to-end workflow proposed by CosmosID underscores the meticulous process involved in COVID-19 metagenomics & metatranscriptomics. From sample collection and RNA extraction to sequencing, data analysis, and visualization, this comprehensive approach ensures a thorough understanding of the viral landscape.
In the pursuit of advancing research and combating future respiratory viral threats, CosmosID invites collaboration with the scientific community. The application of their analysis platform, with real-time bioinformatics capabilities and an intuitive graphical interface, stands as a valuable resource for researchers seeking rapid and accurate insights into microbial communities associated with COVID-19.
In conclusion, the scientific imperative of COVID-19 metagenomics and metatranscriptomics persists as we head into 2024, emphasizing its crucial role in unraveling the intricacies of the SARS-CoV-2 virus.
Through collaborative efforts and advanced metagenomic and metatranscriptomic analysis, the global scientific community endeavors to enhance our understanding of the virus, ultimately contributing to more effective strategies for disease management and prevention.
From discovery to diagnostics and beyond, CosmosID is your go-to resource for strain-level analysis. Learn how our comprehensive suite of bioinformatics tools, paired with an intuitive graphical interface, can help you get the insights you need on microbial communities associated with COVID-19.
Unlock the power of the microbiome with CosmosID. Get in touch today.
References
BAL Metagenomic Samples: Wuhan Institute of Virology, Chinese Academy of Sciences, Zheng Shi; 2020-02-11 https://www.ncbi.nlm.nih.gov/biosample/SAMN1408219
Zhou, et al. (2020) Nature https://www.nature.com/articles/s41586-020-2012-7
Gralinksi, et al. (2020) Viruses https://www.mdpi.com/1999-4915/12/2/135/htm
Centers for Disease Control and Prevention https://tools.cdc.gov/medialibrary/index.aspx#/microsite/id/403323
Illumina (2017). TruSeq Stranded Total RNA Reference Guide. Accessed February 13, 2020.
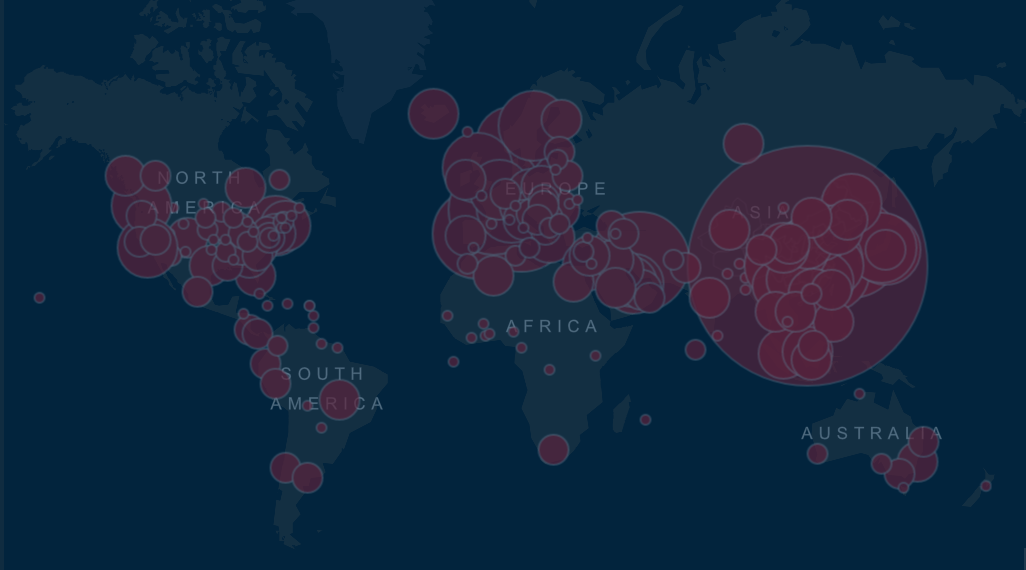
Image Credit: Coronavirus COVID-19 Global Cases by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University: https://gisanddata.maps.arcgis.com/apps/opsdashboard/index.html#/bda7594740fd40299423467b48e9ecf6
