
Introduction
The advent of metagenomic sequencing has led to significant advancements in our understanding of the microbiome in a wide variety of contexts, from the human body1 to farm animals2,3, soil, and marine environments4. Each unique environment provides its own set of challenges in accurate microbial identification, including low microbial biomass in ocean samples, PCR inhibitors in soil samples, and high host DNA in certain human and animal samples. Sequencing of the 16S rRNA gene has been a popular and low-cost way to identify microbes in these samples. However, this method is limited by PCR primer and amplification bias along with unreliable identification below the genus level5. 16S rRNA gene sequencing studies that use primer sets targeting different variable regions cannot be compared directly as different regions selectively detect different bacterial taxa6. Further, relative abundance measures are inaccurate due to variation in the number of 16S rRNA gene operons present within differing bacterial species7.
Whole genome shotgun sequencing (WGS) addresses the amplification, primer bias, and relative quantification issues by avoiding amplification altogether and sequencing the entire bacterial genome. Here, unique, single-copy marker gene sequences or reference genome sequences can be used to identify and quantify bacteria present within a sample more accurately. Because WGS relies on marker genes or alignment to a reference, it is possible to accurately identify microorganisms at the species or even strain taxonomic levels. However, there is wide variation in the accuracy of computational tools developed to perform these microbial identifications. Challenges faced in WGS methods include the requirement to account for variations in genetic diversity within species (i.e. some species are very diverse, whereas others are genetically uniform), mobile elements that are shared among species, the quality of reference genomes used, and the divergence of strains found in nature from the reference genomes that are used for identification. Here, we perform a benchmarking study to evaluate the performance of CosmosID-HUB as compared to five other publicly available taxonomic classification algorithms; Centrifuge11, Metaphlan312, Kraken2_Bracken13, mOTUs229 and Metalign30. These publicly available taxonomic classification algorithms are known for their high accuracy and precision when compared to other publicly available methods based on previous benchmarking evaluations8 0.An ideal metagenomics classifier will properly identify a large number of microorganisms while displaying a small number of false positives at all taxonomic levels. For this study, w e used publicly available benchmarking datasets from CAMI2 (Mouse Gut Dataset)27 and McIntyre et al 2017 benchmarking paper28. The CAMI2 dataset was designed for use as a common benchmarking tool in order to evaluate metagenomics pipelines in a standardized way and the McIntyre datasets consist of mock communities of known compositions.
Importance of Strain Level Resolution
As metagenomics is increasingly becoming a method of choice across multi-disciplinary applications, the importance of sub-species and strain level variation is becoming ever more apparent.14-23 For example, specific strains of Streptococcus mutans produce hemorrhagic damage in the murine brain and other tissues18, whereas other strains are risk factors for ulcerative colitis.17 Likewise, different strains of the protozoan parasite Toxoplasma gondii manifest diverse pathologies and elicit altered host responses19. Particular variants of Staphylococcus epidermidis15 and Staphylococcus aureus16 affect virulence and biofilm formation. Certain strains of Bifidobacterium longum, but not others, protect against pathogens like Escherichia coli, and still others elicit differential immunomodulatory properties.14 Similarly, strain-specific immunomodulatory effects are seen for Propionibacterium freudenreichii21 and for another probiotic agent, Lactobacillus casei, variants derived from different ecological niches vary in their ability to bind foodborne carcinogens.22 The importance of strain resolution is much more apparent when assigning attribution, as exemplified in outbreaks of nosocomial infections such as Legionella pneumophila23-24 and Klebsiella pneumoniae.25 These examples serve to underscore why sub-species and strain level identification is so crucial to our understanding of microbial symbiosis and dysbiosis, and thus demonstrate the power of CosmosID-HUB metagenomics in defining the microbiome composition at a finer taxonomic resolution – critical information needed in microbiome research, epidemiological studies, microbial forensics, and outbreak investigations.
Evaluation of the CosmosID-HUB Taxonomic Profiler
In this study, we compared the performance of the CosmosID-HUB taxonomic profiling algorithm (CosmosID-HUB), to that of Centrifuge11, Metaphlan312, Kraken2_Bracken13, mOTUs229 and Metalign30.
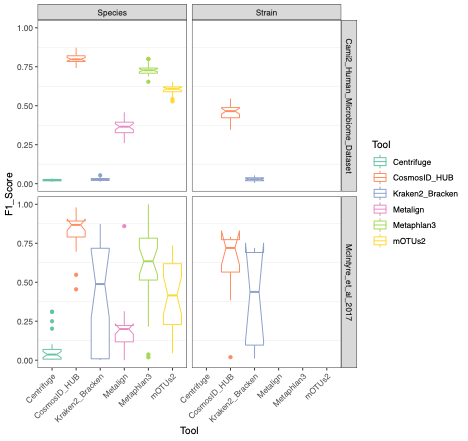
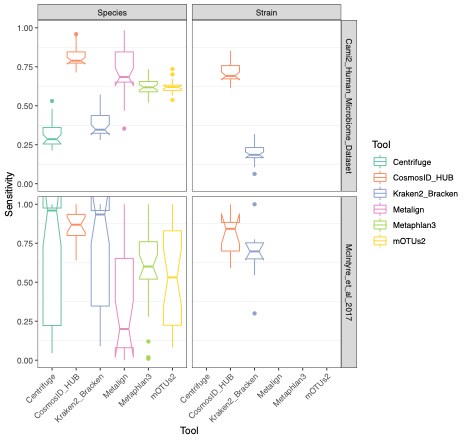
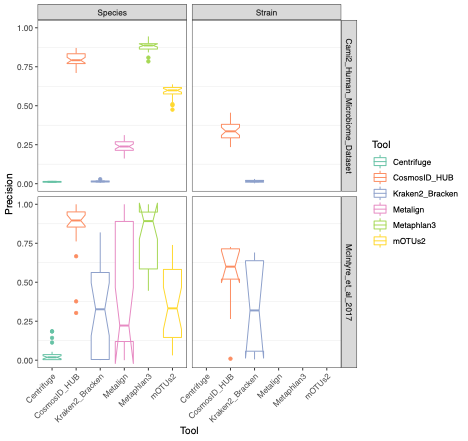
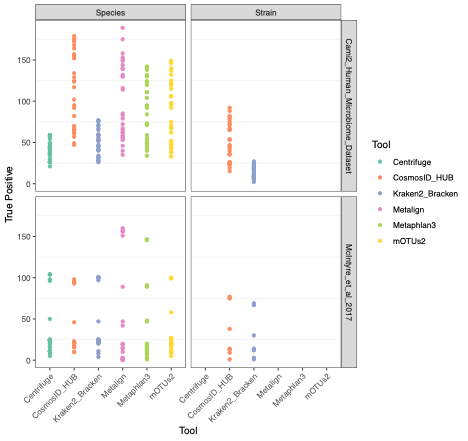
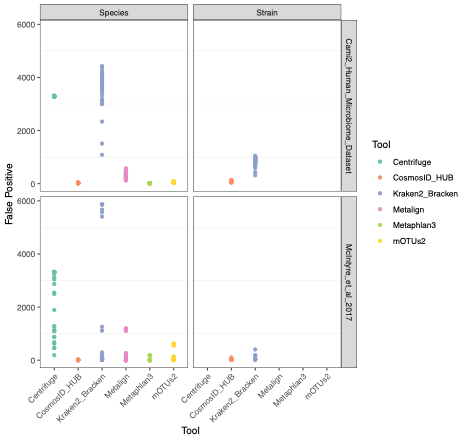
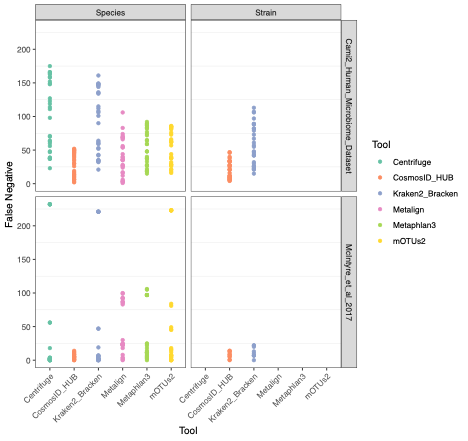
To evaluate the performance of these tools, we used datasets of known microbial composition from CAMI2 and McIntyre et al 2017 benchmarking papers respectively to determine true positives and negatives and false positives and negatives from each of the pipelines. While tools can produce either very aggressive or highly conservative predictions of community composition, to be reliably used in multi-disciplinary microbiome applications it is critical that overall classification accuracy and detection resolution of a tool maintain low rates of false positives and false negatives. Therefore, we evaluated the recall or sensitivity of the results (fraction of species actually present in the metagenomes that are correctly detected) and the precision (fraction of species identified that were actually included in the mock community). Since there is often a trade-off between precision and sensitivity, we also calculated the F1 score, which is the harmonic mean of sensitivity and precision which helps to evaluate both metrics in one score. In figure 1, the performance of the evaluated tools is compared at different taxonomic levels. CosmosID-HUB had the highest F1 score, outperforming the other tools at both species and strain levels. On recall at species level (Figure 2), Kraken2_Bracken and Centrifuge outperformed CosmosID-HUB for the McIntyre et al 2017 benchmarking dataset, but at the cost of a very high number of false positives (Figure 5). This is reflected in the precision metric (Figure 3), where both Kraken2_Bracken and Centrifuge performed poorly. Similarly, Metaphlan3 performed better on precision but at the cost of recall. Since CosmosID-HUB’s performance across precision and recall was similar, the F1 score clearly reflects CosmosID-HUB’s superior performance in correctly identifying the right taxa in the dataset while keeping the false positives low. Lastly, at strain level resolution, CosmosID-HUB was clearly superior and outperformed Kraken2_Bracken across F1 score, precision, and recall. Centrifuge, Metaphlan3 Metalign and mOTUs2 were unable to make strain-level calls.
The public tools that are used in this evaluation fall into two different categories; 1) DNA to DNA based methods (Kraken2_Bracken, Centrifuge, and Metalign) that compare sequencing reads with an exhaustive collection of whole prokaryotic genomes and 2) DNA to marker gene based methods (Metaphlan3 and mOTUs2) that query only specific gene families or clade specific biomarkers in their reference databases to the sequencing reads. Comparison of the sequencing reads to the entire genome allows discrimination among different strains within a certain species. However, it is extremely challenging to carry out strain level analysis primarily because of short reads mapping to multiple genomes due to either local or global homology between different species and within species as well. This is why only Kraken2_Bracken was able to go down to strain level whereas the rest of the DNA to DNA methods (Centrifuge and Metalign) were unable to. The DNA to marker gene based methods are unable to go down to strain level resolution since their database is structured in a manner that consists of specific marker genes per clade rather than comparison to the entire genome. Furthermore, in recent years, the authors of Metaphlan3 have developed a companion tool called StrainPhlAn12 which utilizes the marker gene sequences in the Metaphlan3 database and analyzes the variants within these markers to genotype the strains of each species. However, StrainPhlAn is limited to genotyping only the most abundant species and the identification of only the most abundant strain per species in a community, since detecting multiple strains per species requires much greater depth (well above 10X). CosmosID-HUB’s unique approach to taxonomic profiling allows it to take full advantage of the entire genome of prokaryotes as well as focus on unique signatures/biomarkers per strain to accurately and precisely discriminate between different strains even at lower coverage than 10X.
Conclusions
Overall, at species and strain levels, CosmosID-HUB performs better than Centrifuge, Kraken2_Bracken, Metaphlan3, Metalign and mOTUs2 across all evaluation metrics and particularly on the combined F1 score (the harmonic mean of sensitivity and precision). It’s important to note here that, except Kraken2_Bracken, all the other remaining tools are unable to identify taxa to the strain level. The primary reason why most of the taxonomic profilers are unable to go down to strain level is because of short reads mapping to multiple genomes due to either local or global homology within the same species and different species as well. CosmosID-HUB’s unique ability to differentiate between core and shared biomarkers among different prokaryotic genomes allows it to discriminate among strains of the same species accurately and precisely.
Future Directions for the CosmosID-HUB Cloud-Based Platform
As the field of microbial genomics advances rapidly, we continue to actively work on new R&D projects to update and improve our pipelines and methods. This includes integration of techniques to increase our taxonomic identification resolution as well as new methods that push the boundary of microbiome data insights. For example, the decreasing cost of WGS sequencing has led to an increase of sequencing data from different specimen types in our pipeline, which gives us the opportunity to understand the complexities and nuances associated with a range of sample types. In order to help researchers get the most out of this data, we are working on creating advanced specimen type-specific filters to further increase our accuracy and precision across the entire spectrum of microbiome sample types. Furthermore, with rapid advancements in long-read sequencing technology, both in terms of cost and error rate, we are also actively working on a dedicated pipeline to support both long read classification and metagenome assembled genomes (MAGs)/genome reconstruction. Lastly, in this age of modern high throughput “omics” technologies ranging from metagenomics and metatranscriptomics to metaproteomics and metabolomics, CosmosID-HUB is dedicated to developing a unifying and comprehensive analysis platform where you can combine and aggregate all these data types with their associated clinical and experimental metadata and turn them into actionable insights for your translational and basic science research. This approach will involve using artificial intelligence, machine learning, advanced statistical models, and co-occurrence networks to integrate and interpret your multi-omics data. To conclude, we remain dedicated to helping you achieve actionable insights from your data using flexible, accurate, and cutting-edge methods that produce the greatest biological relevance and meaning.
How to Cite Us and Algorithm Appendix:
Please refer to our Literature web page to cite the previous peer-reviewed publications that have used the CosmosID-HUB Taxonomic Profiling algorithm for their microbiome related research.
The CosmosID-HUB taxonomic profiling algorithm has two separable comparators: the first consists of a pre-computation phase for the reference database and a per-sample computation. The input to the pre-computation phase is a comprehensive curated collection of reference microbial genomes and its output is a phylogeny tree, together with sets of variable length k-mer fingerprints (biomarkers) that are uniquely identified with distinct nodes, branches and leaves of the tree.
The second per-sample, computational phase searches the hundreds of millions of short sequence reads or contigs from draft assembly against the k-mer fingerprint sets. The resulting statistics are analyzed to give fine-grain composition and relative abundance estimates. The second comparator uses edit distance-scoring techniques to compare a target genome with a reference set. Overall classification precision is maintained through aggregation statistics. Enhanced detection specificity is achieved by running the comparators in sequence.
The first comparator finds reads in which there is an exact match with a k-mer uniquely identified with a reference genome; the second comparator then statistically scores the entire read against the reference to verify that the read is indeed uniquely identified with that reference. High-performance bioinformatics enables CosmosID to deliver speed and accuracy in microbial identification. The omprehensive curation collection of reference microbial genomes structured in a unique tree-like database structure provide extremely fine resolution in identification, discrimination of pathogens from ‘near-neighbours’, and accurate measurement of relative abundance. The user-friendly and secure cloud interface offers simplicity in operation, ease of use (without requiring bioinformatics skills), built-in multivariate comparative analysis and advanced statistical analysis modules to allow users to gain informative insights from their metagenomics datasets.
Methods
Dataset Sources
Publicly available metagenomics benchmarking datasets from CAMI227 and McIntyre et al 201728 paper which consisted of mock communities of known compositions were used for this study.
Sample Processing Through Each Pipeline
The dataset has been ran through Centrifuge13, Metaphlan314, Metalign29, mOTUs230, and Kraken2_Bracken15 with default databases. For the ComosID_HUB Microbiome, the dataset was run through the Bacteria Beta 2.1.0 workflow available on CosmosID-HUB cloud platform.
Result files were automatically grouped by connecting the ground truth files to the corresponding output files from Centrifuge, Metaphlan3, Metalign, mOTUs2, Kraken2_Bracken and CosmosID-HUB Microbiome. The ground truth files in tsv format provided the NCBI Taxonomy IDs for species, and strain level per call. The result files were then parsed into python dictionaries and sets accordingly for downstream use.
Downloading NCBI Taxonomy Browser Database
NCBI Taxonomy Browser Database files were downloaded using wget through NCBI’s File Transfer Protocol (FTP) site. Nodes.dmp was parsed into a python dictionary to allow quick identification of Taxonomy Rank given an NCBI Taxonomy ID.
Defining and Calculating Base Statistics
Python 2.7 set operations are noncommutative and were leveraged in the calculation of base statistics. Our base statistics are True Positives, False Positives, True Negatives, False Negatives are defined as follows:
True Positive (TP) – For a given tool, a call is a true positive if a corresponding entry is in the ground truth. True positives were calculated by taking the size of a resultant set. The resultant set was calculated through the intersection of a truth and result set of NCBI Taxonomy IDs.
False Positive (FP) – For a given tool, a call is a false positive if a corresponding entry is not in the ground truth. False positives were calculated by taking the size of a resultant set. The false positives were found by calculating the set difference of the result set and truth set.
False Negative (FN) – For a given tool, a false negative exists if no call is made for a corresponding entry in the ground truth. False negatives were calculated by taking the size of a resultant set. The false negatives were found by calculating the set difference of the truth set and result set.
True Negative (TN) – True Negatives were calculated by subtracting the calls made (TP + FP) from the total number of possible calls for a tool and taxonomy rank being evaluated. The total number of possible calls was calculated by parsing the DB files for a given tool and use of the Taxonomy browser dictionary where appropriate.
Supplementary Statistics
The calculation of our supplementary statistics were dependent on base statistic values.
Our supplementary statistics were F1, Sensitivity, and Precision were calculated with the following formulas:
Sensitivity: TPR=TP/(TP+FN)
Precision: PPV=TP/(TP+FP)
F1Score: F1=2TP/(2TP+FP+FN)
References
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207-14.
- Kauter A, Epping L, Semmler T, Antao E-M, Kannapin D, Stoeckle SD, Gehlen H, Lübke-Becker A, Günther S, Wieler LH, Walther B. The gut microbiome of horses: current research on equine enteral microbiota and future perspectives. Anim Microbiome. 2019;1(1):14.
- Bergamaschi M, Tiezzi F, Howard J, Huang YJ, Gray KA, Schillebeeckx C, McNulty NP, Maltecca C. Gut microbiome composition differences among breeds impact feed efficiency in swine. Microbiome. 2020;8(1):110.
- Logares R, Deutschmann IM, Junger PC, Giner CR, Krabberød AK, Schmidt TSB, Rubinat-Ripoll L, Mestre M, Salazar G, Ruiz-González C, Sebastián M, de Vargas C, Acinas SG, Duarte CM, Gasol JM, Massana R. Disentangling the mechanisms shaping the surface ocean microbiota. Microbiome. 2020;8(1):55.
- Johnson JS, Spakowicz DJ, Hong B-Y, Petersen LM, Demkowicz P, Chen L, Leopold SR, Hanson BM, Agresta HO, Gerstein M, Sodergren E, Weinstock GM. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat Commun. 2019;10(1):5029.
- Chakravorty S, Helb D, Burday M, Connell N, Alland D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods. 2007;69(2):330-9.
- Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF. Divergence and redundancy of 16S rRNA sequences in genomes with multiple rrn operons. J Bacteriol. 2004;186(9):2629-35.
- Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, Gregor I, Majda S, Fiedler J, Dahms E, Bremges A, Fritz A, Garrido-Oter R, Jørgensen TS, Shapiro N, Blood PD, Gurevich A, Bai Y, Turaev D, DeMaere MZ, Chikhi R, Nagarajan N, Quince C, Meyer F, Balvočiūtė M, Hansen LH, Sørensen SJ, Chia BKH, Denis B, Froula JL, Wang Z, Egan R, Don Kang D, Cook JJ, Deltel C, Beckstette M, Lemaitre C, Peterlongo P, Rizk G, Lavenier D, Wu Y-W, Singer SW, Jain C, Strous M, Klingenberg H, Meinicke P, Barton MD, Lingner T, Lin H-H, Liao Y-C, Silva GGZ, Cuevas DA, Edwards RA, Saha S, Piro VC, Renard BY, Pop M, Klenk H-P, Göker M, Kyrpides NC, Woyke T, Vorholt JA, Schulze-Lefert P, Rubin EM, Darling AE, Rattei T, McHardy AC. Critical Assessment of Metagenome Interpretation-a benchmark of metagenomics software. Nat Methods. 2017;14(11):1063-71.
- Ye SH, Siddle KJ, Park DJ, Sabeti PC. Benchmarking metagenomics tools for taxonomic classification. Cell. 2019;178(4):779-94.
- Meyer F, Bremges A, Belmann P, Janssen S, McHardy AC, Koslicki D. Assessing taxonomic metagenome profilers with OPAL. Genome Biol. 2019;20(1):51.
- Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26(12):1721-9.
- Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, Valles-Colomer M, Weingart G, Zhang Y, Zolfo M, Huttenhower C, Franzosa EA, Segata N. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife. 2021;10.
- Wood DE, Lu J, Langmead B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019;20(1):257.
- Fukuda S, Toh H, Hase K, Oshima K, Nakanishi Y, Yoshimura K, Tobe T, Clarke JM, Topping DL, Suzuki T, Taylor TD, Itoh K, Kikuchi J, Morita H, Hattori M, Ohno H. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469(7331):543-7.
- Gill SR, Fouts DE, Archer GL, Mongodin EF, Deboy RT, Ravel J, Paulsen IT, Kolonay JF, Brinkac L, Beanan M, Dodson RJ, Daugherty SC, Madupu R, Angiuoli SV, Durkin AS, Haft DH, Vamathevan J, Khouri H, Utterback T, Lee C, Dimitrov G, Jiang L, Qin H, Weidman J, Tran K, Kang K, Hance IR, Nelson KE, Fraser CM. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187(7):2426-38.
- Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010;465(7296):346-9.
- Kojima A, Nakano K, Wada K, Takahashi H, Katayama K, Yoneda M, Higurashi T, Nomura R, Hokamura K, Muranaka Y, Matsuhashi N, Umemura K, Kamisaki Y, Nakajima A, Ooshima T. Infection of specific strains of Streptococcus mutans, oral bacteria, confers a risk of ulcerative colitis. Sci Rep. 2012;2:332.
- Nakano K, Hokamura K, Taniguchi N, Wada K, Kudo C, Nomura R, Kojima A, Naka S, Muranaka Y, Thura M, Nakajima A, Masuda K, Nakagawa I, Speziale P, Shimada N, Amano A, Kamisaki Y, Tanaka T, Umemura K, Ooshima T. The collagen-binding protein of Streptococcus mutans is involved in haemorrhagic stroke. Nat Commun. 2011;2:485.
- Saeij JPJ, Boyle JP, Boothroyd JC. Differences among the three major strains of Toxoplasma gondii and their specific interactions with the infected host. Trends Parasitol. 2005;21(10):476-81.
- Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF. Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res. 2013;23(1):111-20.
- Foligné B, Deutsch S-M, Breton J, Cousin FJ, Dewulf J, Samson M, Pot B, Jan G. Promising immunomodulatory effects of selected strains of dairy propionibacteria as evidenced in vitro and in vivo. Appl Environ Microbiol. 2010;76(24):8259-64.
- Hernandez-Mendoza A, Garcia HS, Steele JL. Screening of Lactobacillus casei strains for their ability to bind aflatoxin B1. Food Chem Toxicol. 2009;47(6):1064-8.
- Helbig JH, Kurtz JB, Pastoris MC, Pelaz C, Lück PC. Antigenic lipopolysaccharide components of Legionella pneumophila recognized by monoclonal antibodies: possibilities and limitations for division of the species into serogroups. J Clin Microbiol. 1997;35(11):2841-5.
- Visca P, Goldoni P, Lück PC, Helbig JH, Cattani L, Giltri G, Bramati S, Castellani Pastoris M. Multiple types of Legionella pneumophila serogroup 6 in a hospital heated-water system associated with sporadic infections. J Clin Microbiol. 1999;37(7):2189-96.
- Snitkin ES, Zelazny AM, Thomas PJ, Stock F, NISC Comparative Sequencing Program Group, Henderson DK, Palmore TN, Segre JA. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4(148):148ra116.
- Marx V. Microbiology: the road to strain-level identification. Nat Methods. 2016;13(5):401-4.
- Meyer F, Fritz A, Deng Z-L, Koslicki D, Lesker TR, Gurevich A, Robertson G, Alser M, Antipov D, Beghini F, Bertrand D, Brito JJ, Brown CT, Buchmann J, Buluç A, Chen B, Chikhi R, Clausen PTLC, Cristian A, Dabrowski PW, Darling AE, Egan R, Eskin E, Georganas E, Goltsman E, Gray MA, Hansen LH, Hofmeyr S, Huang P, Irber L, Jia H, Jørgensen TS, Kieser SD, Klemetsen T, Kola A, Kolmogorov M, Korobeynikov A, Kwan J, LaPierre N, Lemaitre C, Li C, Limasset A, Malcher-Miranda F, Mangul S, Marcelino VR, Marchet C, Marijon P, Meleshko D, Mende DR, Milanese A, Nagarajan N, Nissen J, Nurk S, Oliker L, Paoli L, Peterlongo P, Piro VC, Porter JS, Rasmussen S, Rees ER, Reinert K, Renard B, Robertsen EM, Rosen GL, Ruscheweyh H-J, Sarwal V, Segata N, Seiler E, Shi L, Sun F, Sunagawa S, Sørensen SJ, Thomas A, Tong C, Trajkovski M, Tremblay J, Uritskiy G, Vicedomini R, Wang Z, Wang Z, Wang Z, Warren A, Willassen NP, Yelick K, You R, Zeller G, Zhao Z, Zhu S, Zhu J, Garrido-Oter R, Gastmeier P, Hacquard S, Häußler S, Khaledi A, Maechler F, Mesny F, Radutoiu S, Schulze-Lefert P, et al. Critical Assessment of Metagenome Interpretation: the second round of challenges. Nat Methods. 2022;19(4):429-40.
- McIntyre ABR, Ounit R, Afshinnekoo E, Prill RJ, Hénaff E, Alexander N, Minot SS, Danko D, Foox J, Ahsanuddin S, Tighe S, Hasan NA, Subramanian P, Moffat K, Levy S, Lonardi S, Greenfield N, Colwell RR, Rosen GL, Mason CE. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017;18(1):182.
- Milanese A, Mende DR, Paoli L, Salazar G, Ruscheweyh H-J, Cuenca M, Hingamp P, Alves R, Costea PI, Coelho LP, Schmidt TSB, Almeida A, Mitchell AL, Finn RD, Huerta-Cepas J, Bork P, Zeller G, Sunagawa S. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat Commun. 2019;10(1):1014.
- LaPierre N, Alser M, Eskin E, Koslicki D, Mangul S. Metalign: efficient alignment-based metagenomic profiling via containment min hash. Genome Biol. 2020;21(1):242.
