

In metagenomics, biological specimens are sometimes collected from host tissue that contains a high ratio of host to bacterial DNA. When shotgun metagenomics sequencing data are generated for these specimens, they normally contain a large fraction of host reads compared to microbial reads. Analyzing microbiome data with a high host ratio of reads can lead to spurious results, especially in the case of functional characterization.
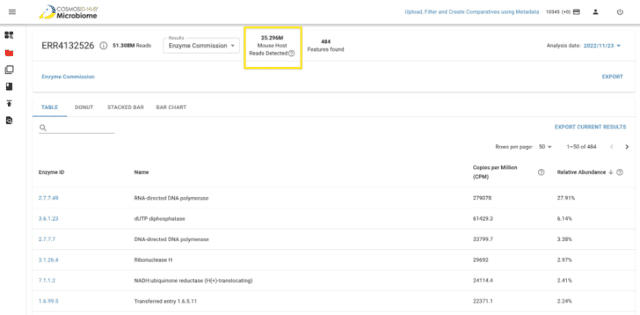
In order to address this issue bioinformatically, we are pleased to introduce a fully automated preprocessing workflow to remove host sequencing reads from your metagenomics sequencing data. Once a user uploads their metagenomics sequencing reads to the HUB, they will be allowed to choose from a list of 8 different host species (Figure 1) to de-host their sequencing reads. The percentage removal statistics for de-hosting will be available both from the user interface and the export as well. More information about our fully automated de-hosting workflow is available within our CosmosID-HUB documentation.
The new release of the Automated De-Hosting workflow is part of CosmosID’s microbial genomics platform, CosmosID-HUB Microbiome, which is continually expanding with various features and workflows for analyzing microbial communities and for inferring translational and actionable microbiome insights.
Currently, CosmosID-HUB Microbiome analyzes 16S, whole genome (shotgun metagenomics and metatranscriptomics), and ITS sequencing data and provides options for discovering antimicrobial resistance (AMR) and virulence markers, microbial taxa (composition), and functions.
Users are also able to aggregate and compare their primary analysis results (taxa, AMR and virulence markers, functions) by cohorts or groups using statistical modules to gain actionable insights from their data.
