
Thank you for your interest in our series of vaginal microbiome comparisons. Based on the results of this month’s survey, today we follow up on the functional metagenomic diversity of the non-pregnant community state type (CST) I female vaginal microbiome.
As a quick refresher, a healthy vaginal microbiome harbors low microbial diversity, as opposed to the more diverse gut microbiome, and is usually dominated by Lactobacillus species. Lactobacillus dominance is so common that vaginal community state types (CST) have been defined (Ravel et al 2011) in which specific Lactobacillus species dominate the community. In brief, L. crispatus dominance defines CST I, L. gasseri dominance defines CST II, L. iners dominance underlines CST III, and L. jensenii dominance marks CST V. There is a final CST IV which is not defined by Lactobacillus dominance but instead by a lack of lactobacilli and increased species diversity. These are all considered healthy vaginal states. For our past comparison elucidating the clustering of healthy vaginal microbiomes from different countries by CSTs, we included vaginal whole-genome sequencing (WGS) samples of women from the USA (n = 51, Mitchell et al 2020), Sweden (n = 45, Sterpu et al 2021) and China (n = 35, Yang et al 2020). Additionally, we included vaginal microbiome data from the USA Human Microbiome Project study (n = 30) as a control. The heatmap based on sample similarity of the non-pregnant vaginal microbiome comparison illustrated that the samples clustered by CSTs, but not by study or country. This suggests that CST was the principal driver of vaginal microbiome similarity.
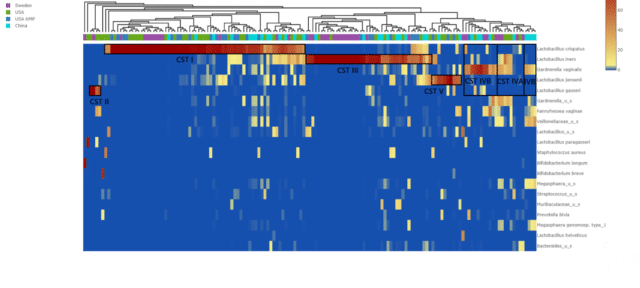
We took a deeper dive into this data to examine whether vaginal CSTs displayed differences in predicted functional pathways. Using metagenomics and metatranscriptomics, a study by France et al. (2022) illustrated that the transcriptional activity of vaginal pathogens varies depending on the community in which they reside, suggesting ecological interactions that may increase or decrease pathogenic potential. Consequently, exploring functional metagenomics within a CST may further illuminate vaginal microbiome biodiversity and have medical implications. To elucidate functional diversity within CSTs, we plotted the heatmap of functional compositions by country (Fig. 2).
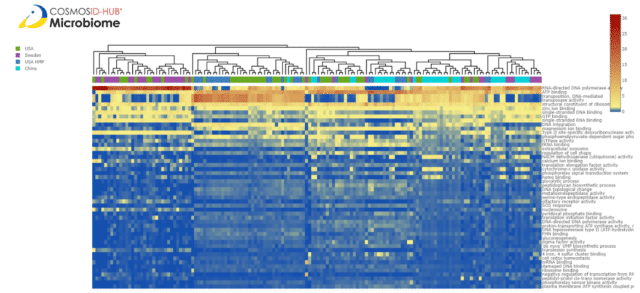
Plotting the heatmap of functional gene ontology (GO) terms relative abundance similarity shows some clustering of the samples by country, but intermixing of the samples suggests some other driver of functional similarity. There are five distinct clusters present but country does explain them. To understand if CST is contributing to similarities in vaginal functional metagenomics, we plotted the same heatmap by CSTs (Fig. 3). Here, we see that two of the clusters are unique to CST I and that the other 3 clusters contain a mix of all the other CSTs. We calculated the Bray-Curtis dissimilarity between these samples and plotted them in Figure 4. Here, all CST I samples cluster distinctly from the other CSTs and possibly create two clusters. We decided to take a deeper dive into these two CST I clusters to determine what functions are differential between them.
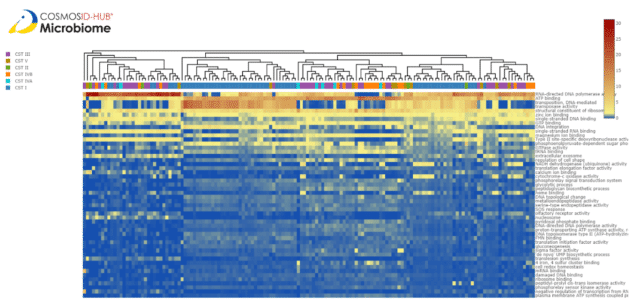
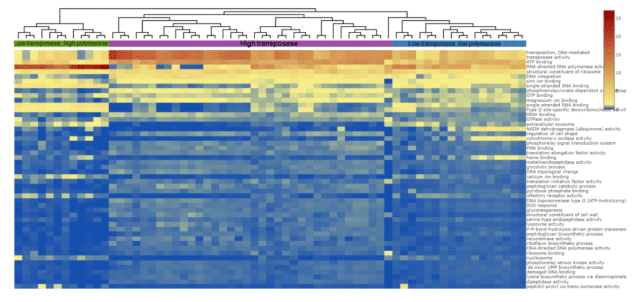
A closer look at the relative abundance of predicted functions in the two CST I clusters suggested that RNA-dependent polymerase and transposase activity may play a role. To understand their impact in divergence, we classified CST I samples as having high and low RNA-dependent polymerase and transposase activity. When labeled this way and plotted again on the heatmap, we saw three clusters within the CST I samples; “High transposase activity”, “Low transposase, high RNA-dependent polymerase activity” and “Low transposase, low RNA-dependent polymerase activity” groups (Fig 5). This suggests that predicted polymerase and transposase activities may create distinct functional groups within CST I. Given that these activities are involved in RNA and DNA creation and movement of genetic sequences, this may imply strain-level differences among CST I lactobacilli.
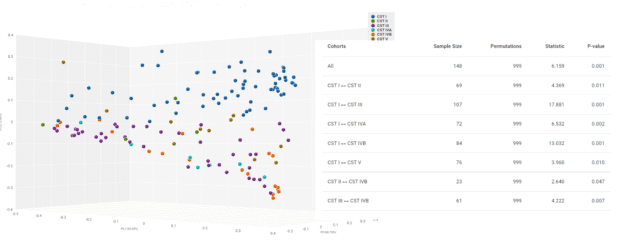
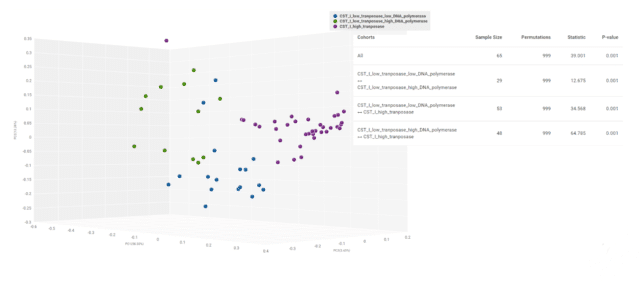
Next, we plotted a 3D visualization of the Bray-Curtis dissimilarity matrix and tested the significance of compositional divergence using a PERMANOVA test (Fig. 6). The results confirmed that vaginal functional metagenomics are significantly dissimilar in overall composition by different levels of transposase and DNA polymerase activity in CST I.
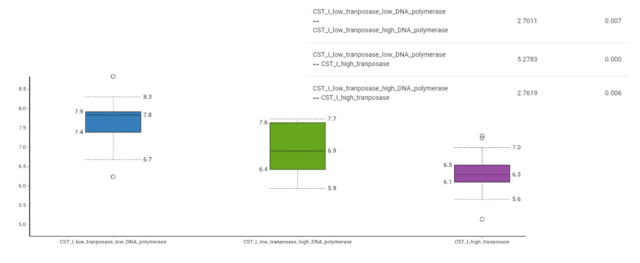
We also calculated alpha diversity between the polymerase and transposase groups using Shannon’s indicex (Fig 7). Significance was assessed using the Wilcoxon Rank Sum Test.
Shannon diversity is significantly higher in the low transposase and DNA polymerase activity group and significantly lower in the high transposase cohort. Shannon’s alpha diversity takes richness, community heterogeneity, and feature evenness (the number of each of those features) into account. This suggests that the low transposase and DNA polymerase activity CST I has the highest number of dominant functional features, meaning it is the most function-rich and the most functionally even. This implies a loss of genes encoding polymerase and transposase activity with increasing functional diversity.
In conclusion, our analysis illustrates the divergence of samples within CST I based on the level of predicted genetic diversity and functional genes responsible for building DNA and RNA sequences and moving them within the genome. A clear limitation of this analysis is our lack of examination of the functional diversity in the other CSTs, which seemed to be functionally intermixed within three different clusters. Understanding functional differences within vaginal CSTs may give insight into nuances within each CST that may play roles in vaginal pathogenicity and disease.
