

Background
Chronic kidney disease is a leading cause of morbidity and mortality around the globe. The disease often leads to kidney failure whe it progresses to end-stage renal disease (ESRD). ESRD produces $34 billion of costs annually to the US economy.
ESRD etiology is often driven by the accumulation of toxic metabolites in body compartments. While many toxins are produced by the human host, evidence suggests that a significant portion of toxic metabolites come from bacteria in the gut microbiome.
ESRD has known impacts on the gut microbiome. Moreover, uremic toxins produced by the gut microbiome can also alter the microbial community composition in patients with chronic kidney disease.
Consequently, investigating the microbiome composition variation in healthy controls and ESRD patients may lead to the identification of biomarkers that could aid in diagnosis and treatment.
Findings
We performed a fecal microbiome comparative analysis of samples from patients with ESRD and healthy controls (original samples from Wang et. al., 2020). We imported the shotgun metagenomic sequencing data from the NCBI Sequence Read Archive (accession code PRJNA449784) into the CosmosID-HUB Microbiome for accurate profiling of the bacterial species’ relative abundances among the samples along with the prediction of functional pathways. Then we compared bacterial relative abundances between ESRD patients and healthy controls.
To compare species-level relative abundances between the two cohorts, we plotted a cohort-clustered species relative abundance heatmap. The heatmap illustrated no clear pattern that distinguished between the gut microbiomes of ESRD patients and healthy controls (Figure 1).
After having found no obvious species-level patterns, we examined if the gut microbiomes of the two cohorts varied at a phylum level. To do that, we plotted aggregated stacked bar plots of phylum-level relative abundances. The barplot illustrated that Actinobacteria is expanded in ESRD patients’ gut microbiome (Figure 2).
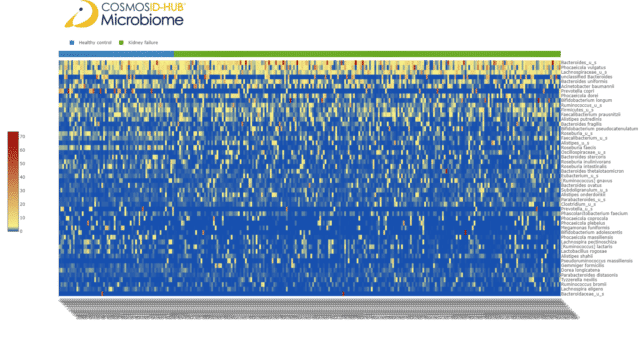
Figure 1: Species-level relative abundance heatmap of ESRD patient and healthy control gut microbiomes
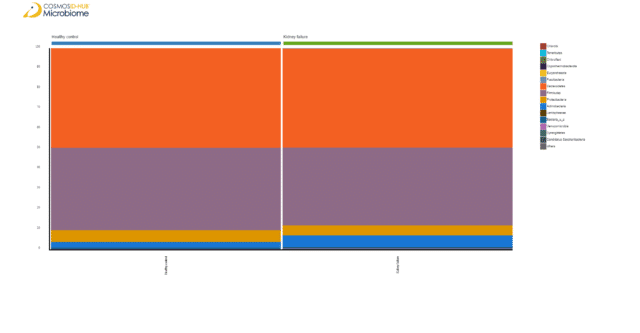
Figure 2: Phylum level relative abundance heatmap of ESRD patient and healthy control gut microbiomes
To examine the variation in species richness and evenness, we calculated the Simpson’s and Shannon’s indices of diversity between the two cohorts using a Wilcoxon rank sum test.
The findings of the Wilcoxon test illustrated a lack of significant differences in alpha diversity with either index (Simpson’s index Wilcoxon Test p-value = 0.923, Shannon index Wilcoxon Test p-value = 0.682, respectively).
The findings of the alpha diversity comparison suggest that gut bacteriomes of ESRD patients and healthy controls host a similar number of different taxa with similar levels of relative abundance distributions.
While bacterial communities may harbor taxa at similar numbers and abundances on a per sample level, they may comparatively be different. In that case, communities with no difference in alpha diversity may be significantly different in beta diversity.
Hence, to determine whether the microbial communities are significantly different in composition between the two cohorts, we calculated the Bray-Curtis dissimilarity matrices between them using the relative abundance of the bacterial species.
We then performed PERMANOVA to test the significance of the dissimilarity result. The PERMANOVA test resulted in a p-value = 0.001, suggesting that the gut bacteriomes of ESRD patients and healthy controls were significantly dissimilar in community composition (Figure 3).
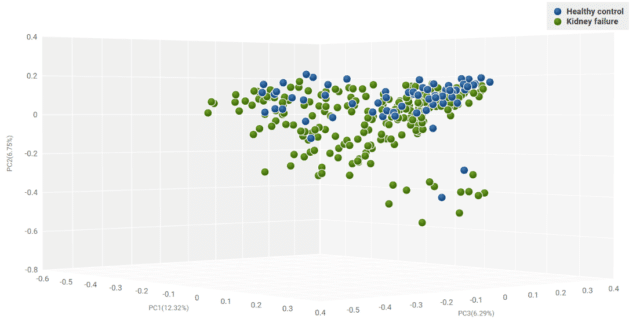
Figure 3: A 3D visualization of Bray-Curtis dissimilarity matrices of gut microbiomes of healthy controls and ESRD patients.
After having observed divergence in the community compositions of the two cohorts, we investigated which taxa may be driving community composition divergence between them. To do that, we performed a linear discriminant analysis of effect size (LefSe) test. We show only taxa with an effect size (LDA score) above 3.5, to illustrate the top discriminative taxa.
According to the LefSe results, Prevotella copri, Roseburia faecis, Megamonas funiformis, Lachnospira pectinoschiza, Acinetobacter baumannii, Lactobacillus rogosae, and Phocaeicola plebeius are significantly enriched in healthy controls, indicating that they were depleted in kidney failure patients.
Conclusions
In conclusion, the gut microbiomes of ESRD patients and healthy controls illustrated similar species richness and evenness. Nonetheless, community compositions emerged to be significantly dissimilar and the depletion of key taxa in the ESRD gut microbiome played a role in community composition divergence.
Those taxa may emerge as biotherapeutic targets to alleviate symptoms if they have causal relationships with metabolism of bacterial toxins.
Altogether, these results illustrate clear microbiome shifts, and could be complemented with a functional metagenomics comparison of the two cohorts to see if functional differences further explain the taxonomic divergence between the two cohorts.
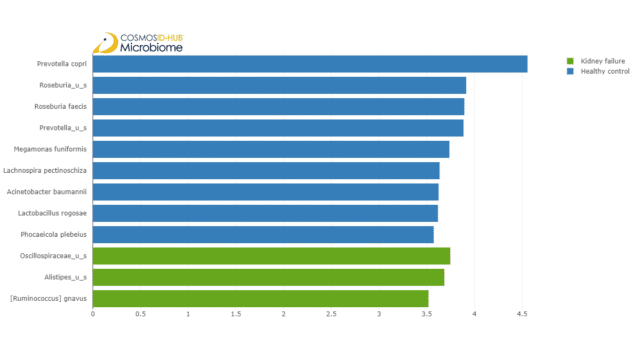
Figure 4: A bar plot illustration of LefSe results
How did the CosmosID-HUB support this research?
CosmosID’s HUB and leading microbiome sequencing services enabled the researchers to gain unprecedented insights into the microbiome shifts between ESRD and healthy control cohorts.
The data from the CosmosID-HUB was used to conduct a Wilcoxon rank sum test, calculate Bray-Curtis dissimilarity matrices, perform PERMANOVA and draw LefSe results for taxonomic and functional differences (Figures 3 & 4).
In short: the CosmosID-HUB is a powerful tool for investigating microbial communities – enabling researchers to gain rapid insights that could otherwise take months or years to uncover.
The cloud-based platform also provides access to advanced analytics tools, allowing researchers to generate meaningful results – and understand them, quickly.
Want results like this? Get started with the CosmosID-HUB today.
Want more like this? Sign-up to our newsletter to get the latest news from CosmosID:
